16S rRNA WORKFLOW REPORT
INTRODUCTION
The 16S rRNA is a ribosomal RNA necessary for the synthesis of all prokaryotic proteins. The genes coding for the 16S rRna has regions that are highly conserved and are shared in all bacteria species making it useful in identifying bacteria in samples. Moreover, the gene has variable regions which can be used to reconstruct phylogenies.
In this miniproject we created two worklows that can be used to determine microbial communities present in different enivironment using the 16S rRNA analysis.
Objectives.
- To develop a 16S-rRNA analysis workflow.
- To test the pipelines developed.
- To analyse the results.
- To compare the tools used.
Input Data Assessment
- The input datasets were of good quality based on the FASTQ results
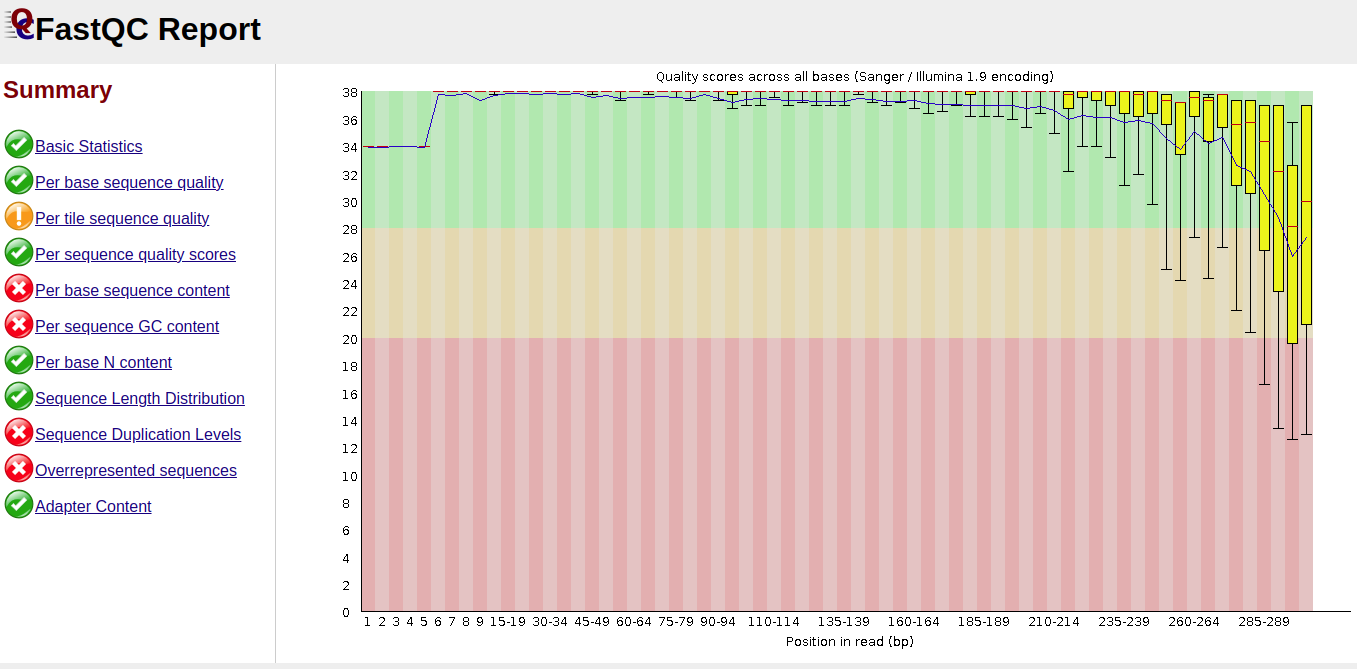
Before Trimmomatic
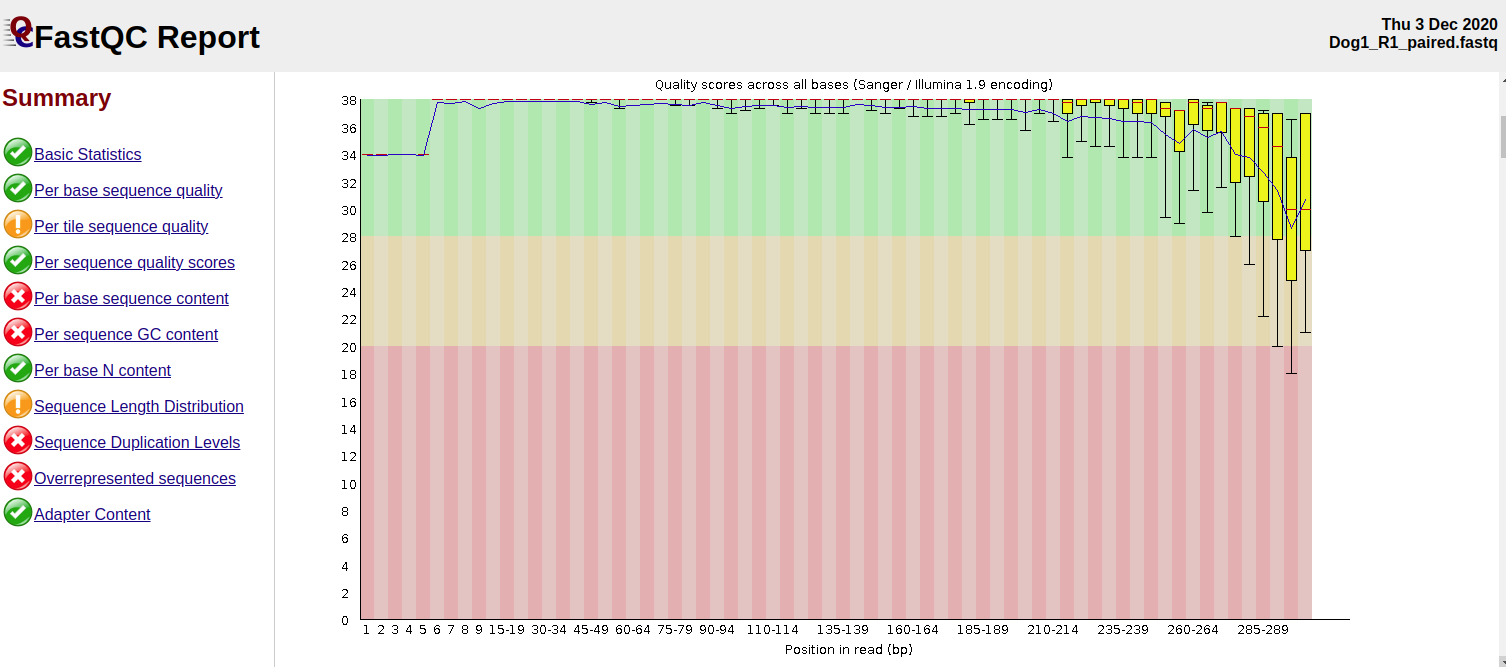
After Trimmomatic
The reads before trimming had an error rate (EE) mean of 3.5 (R2) compared to forward reads which had an E.E mean of about 0.5. The error rate mean reduced to 0.2 for R1 and 0.7 for R2.
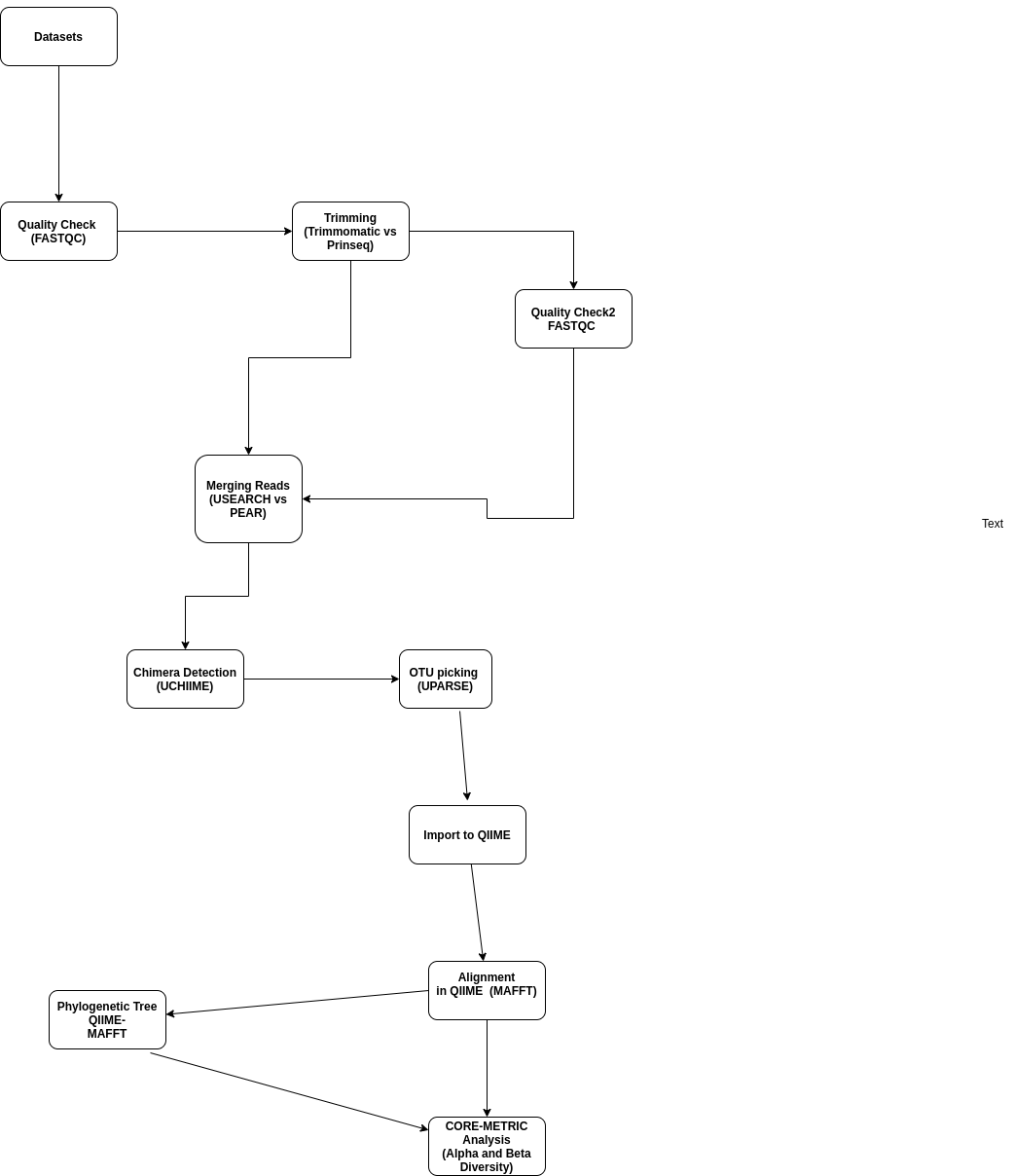
The flow charts below show tools used on the workflows in three phases namely:
- Preprocessing Reads.
- OTU picking,Classification and Phylogenetic Tree Generation.
- Measure Diversity and other Statistical Analyses.
QIIME Workflow

Runtime Analysis
- Time Taken: 106mins
- Hardware Usage- Fair ,user can perform other light tasks while running.
- Storage - 8.9 G.B
Multiple-Tools Workflow
Runtime Analysis
- Time Taken - 2.5 hrs.
- Hardware Usage - Intensive,computer freezes.
- Storage - 8.8 G.B
Operational Assessment
Factors considered
- Speed
- Accuracy
- Cost
- Output Formats and Reports
- Hardware Requirements
Alternative Tools Critique
- Trimmomatic vs Prinseq: Trimmomatic improved quality after trimming but prinseq did not trim anything.
- UPARSE (98% merged) vs PEAR(99% merged): UPARSE does not require input of reverse reads unlike PEAR. PEAR produces 4 outputs.
- UCHIME vs Chimeraslayer: Uchime is faster and detects more chimeras than Chimeraslayer which also requires different formats.
- UPARSE vs QIIME : Qiime picked 209 OTUS while UPARSE had 190 OTUS.DADA2 pipeline picked 314 ASVs.
- UCLUST vs QIIME: UCLUST uses less time than QIIME. We opted to use UPARSE
- QIIME vs Mothur : Mothur deletes all reads in the preparation stage whereas QIIME picked 209 OTUS using deblur plugin. Mothur Tutorial
- MAFFT vs pyNAST for Alignment: Couldnt download pyNAST cause it requires python version 2.7 and below.
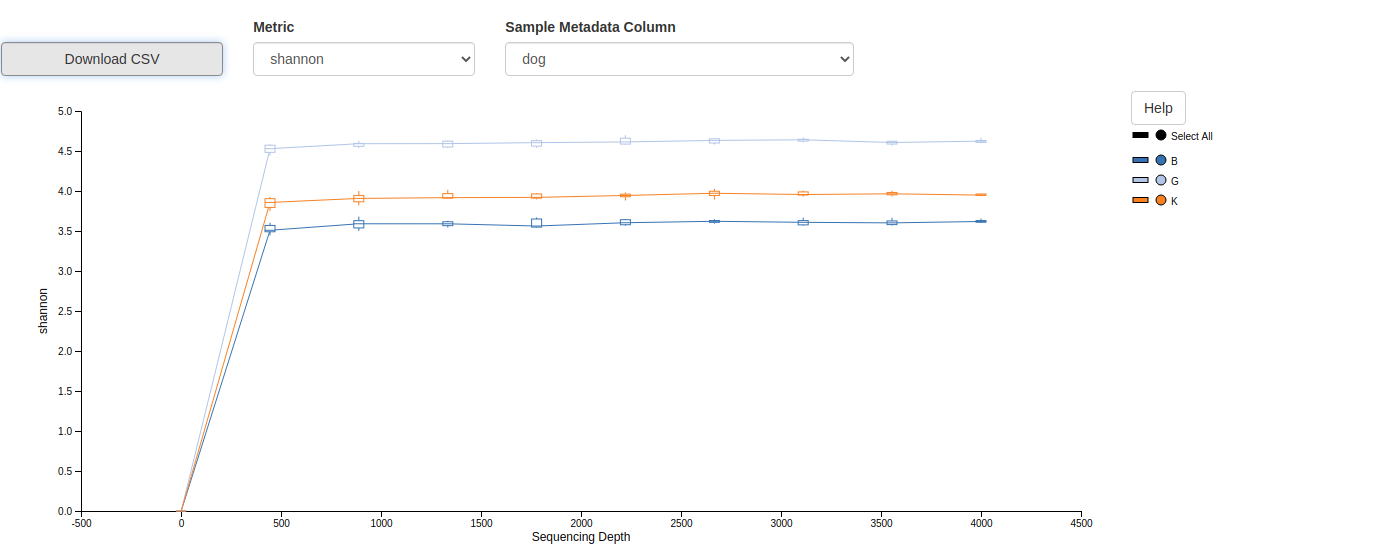
Results Analysis/Metrics Comparions
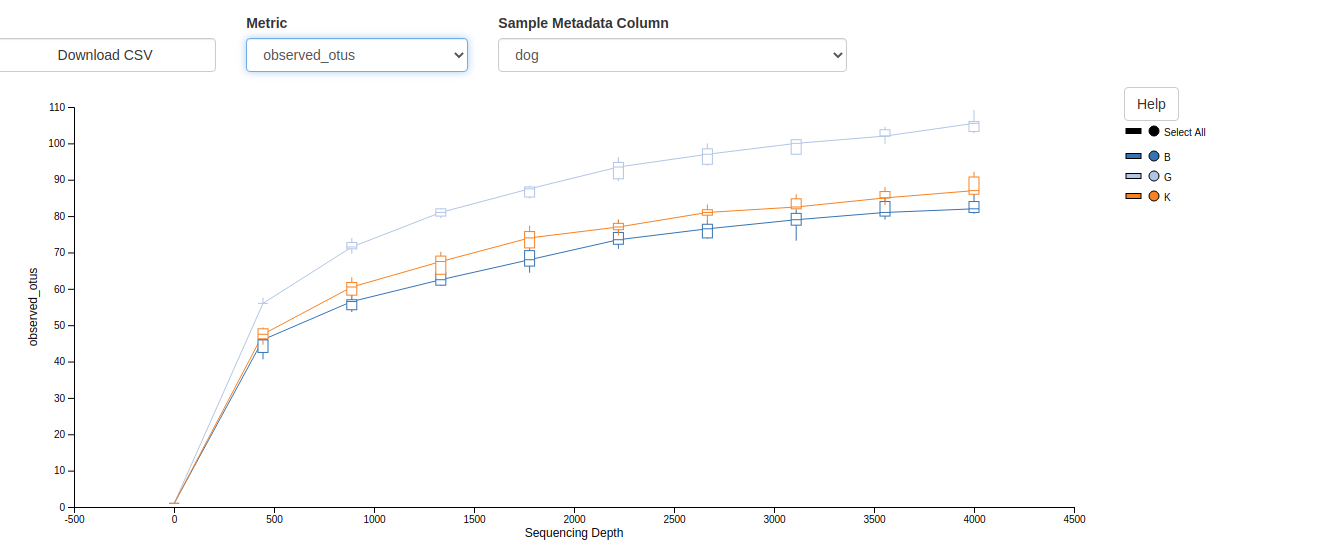
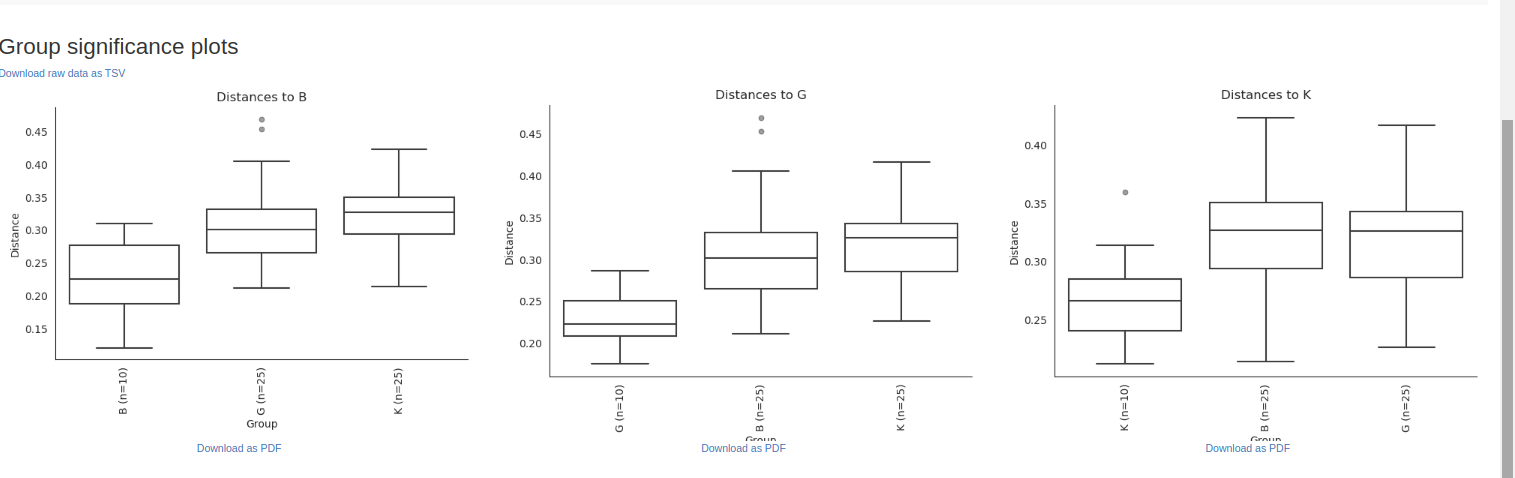
Comparison of the Alpha and Beta diversity analyses from the two workflows.
.svg)
.svg)
.png)
.png)
Other Images from Qiime workflow....
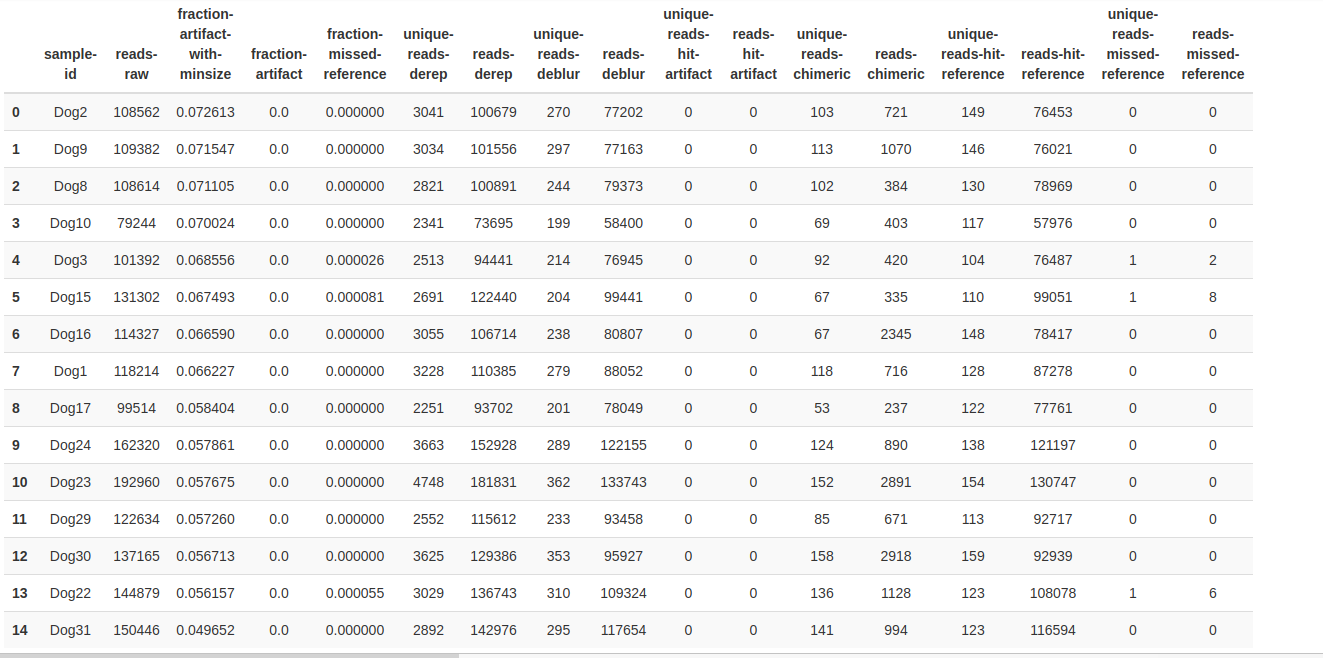
Taxonomy Barplot Alpha rarefaction/Shannon Alpha rarefaction/Observed otus Deblur Stats Rooted Tree Unrooted Tree Circular Tree Taxonomy Table Unifrac Unweighted Unweighted unifrac/Emperor.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
.svg){kind=link}
{kind=link}
{kind=link}
.png){kind=link}